
As a focal point in current life science research, genetic mutation enjoys rapidly evolving detection methodologies synonymous with the field. The advancements in genetic mutation detection techniques hold significant meaning for early disease diagnosis and treatment. The aim of this paper is to provide a concise exposition of various commonly used genetic mutation detection methodologies and their underlying principles. This is intended to aid in appropriate selection based on testing objectives and experimental conditions.
Southern blot
Conceived by the eminent British biologist Edwin Southern in 1975, Southern blot hybridization is a pioneering technique developed for the specific detection of DNA. Anchored in the fundamental principle of base complementarity, this method exploits the precise and selective hybridization of two nucleic acid single strands exhibiting homology under certain conditions, leading to the formation of a double-stranded structure. As the first embodiment of blotting methodologies, Southern blot hybridization maintains a vital presence in the field of molecular biology.
At its core, the mechanism of Southern blot hybridization is founded on the principle of complementary base pairing. This process involves nucleic acid strands, which, possessing a certain degree of homology, selectively participate in the formation of double-stranded structures under predetermined conditions. Provided that the target material incorporates sequences complementary to the probe, their interaction is enabled through precise base pairing. Upon the removal of unbound probes through washing processes, the identification and quantification of the hybridized fragments are achieved using modalities such as autoradiography (Figure 1) or other apposite methodologies.
In light of its superior sensitivity and specificity, Southern blot hybridization rapidly gained recognition as the archetypal molecular detection method in the spectrum of probe hybridization techniques. Its utility spans across myriad fields, such as the delineation of gene mutations and the scrutiny of Restriction Fragment Length Polymorphism (RFLP).
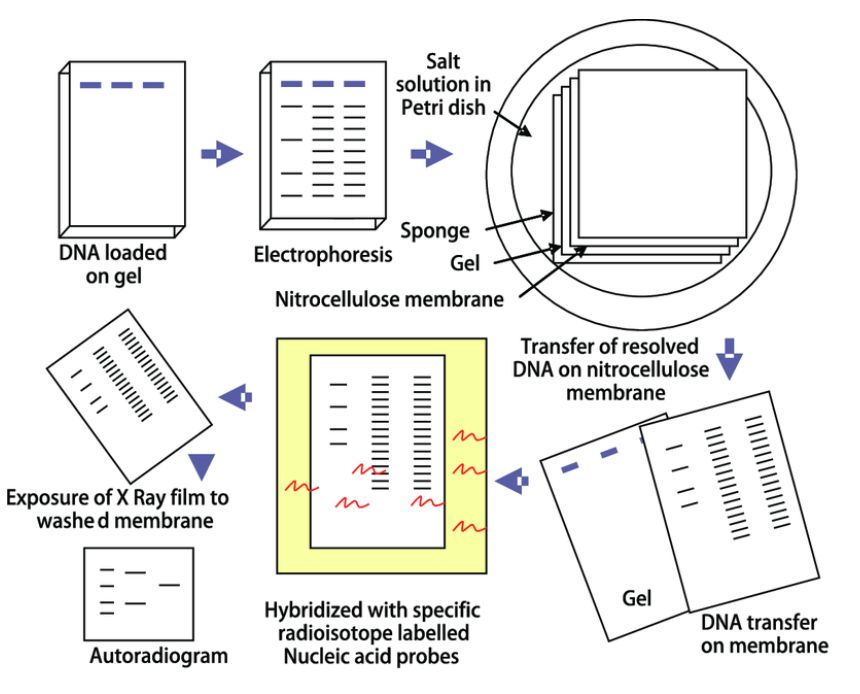
Figure.1 Southern blot analysis (Rajiv Kumar et al.,2012).
PCR
In the domain of gene mutation detection, Southern blot technology has held a prominent position since 1985, capable of discerning various mutation forms such as gene deletions, insertions, and frameshift recombinations. However, under the technological constraints of that era, the inability to artificially amplify target genes within samples rendered certain mutations undetectable by Southern blot. Consequently, mutations eluding Southern blot scrutiny necessitated intricate in vitro oligonucleotide-mediated DNA mutagenesis, followed by laborious DNA sequence analysis for confirmation—a method characterized by its complexity, time-consuming nature, and elevated costs.
The inception of Polymerase Chain Reaction (PCR) technology heralded a paradigm shift in molecular diagnostics, particularly concerning gene mutation detection. Essentially, all modern molecular diagnostic methods pivoted around the PCR methodology. The implementation of PCR technology effectuated substantial advancements in multiple dimensions of detection: it automatized procedures, condensed the timescale of analyses, and significantly bolstered result precision. By all means, the integration of PCR technology constituted a seminal development in gene mutation detection research.
1. ARMS-PCR
The Amplification Refractory Mutation System- Polymerase Chain Reaction (ARMS-PCR), also known as Allele-Specific PCR (AS-PCR), rests upon the principle of allele-specific extension reaction. This technique only proceeds when a specific allele's primer pairs align with the mutation site, allowing the extension reaction to occur. Globally, ARMS-PCR has emerged as one of the most critical and advanced technologies within the realm of individual molecular detection for tumors. Simultaneously, it stands as one of the most accurate technological instruments in the field of genetic disease detection.
The distinction between ARMS-PCR and conventional PCR lies in the design of the primer for allele-specific genes. Two upstream primers have differing 3' end sequences. During the PCR amplification process, upstream primers that cannot fully match the template fail to form completely complementary base pairs, leading to mismatching. Once this mismatching reaches a certain level, primer extension is terminated, and no PCR amplification product of specific length can be obtained, indicating that the template DNA does not have a base pair with the 3' end of the primer, and vice versa (Peng et al.,2018). At present, scientists have extended the ARMS-PCR methodology to incorporate four primers (Tetra-Primer ARMS-PCR), whereby four PCR primers are designed simultaneously, two of which are located on the 3' end SNP site and have opposite directions belonging to different genotypes, while the other two are located outside the SNP (Figure 2).
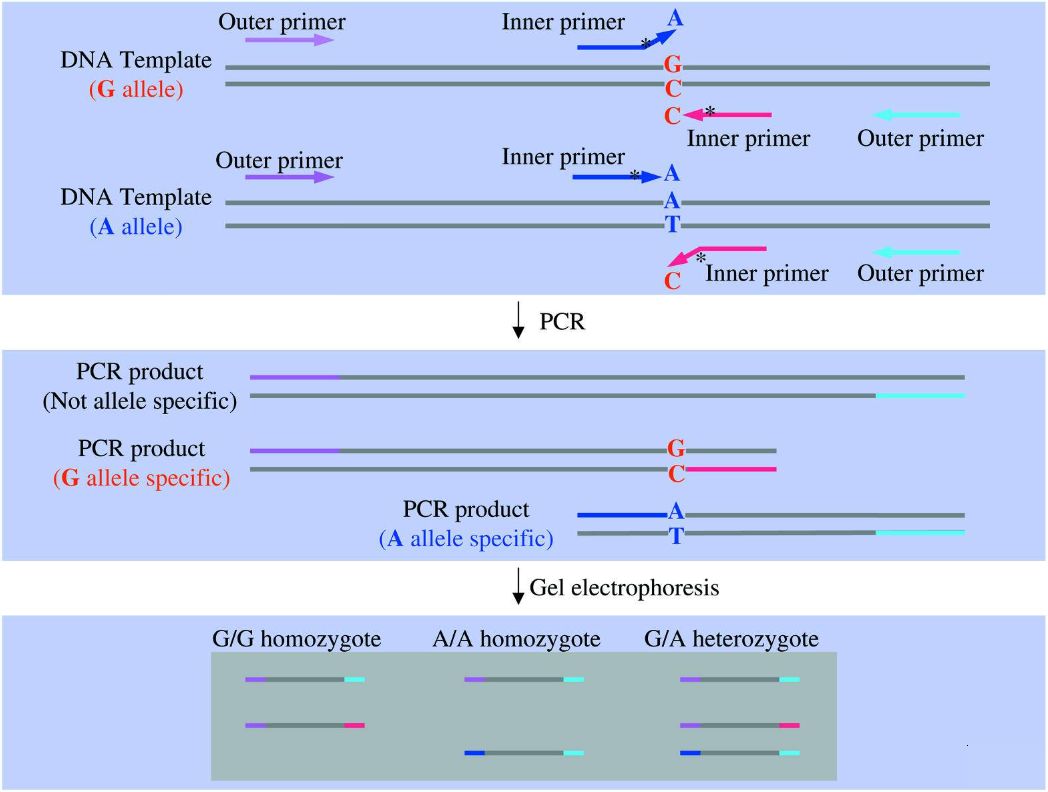
Figure.2 ARMS-PCR (Ye et al., 2001)
2. Digital PCR
Digital PCR (dPCR) epitomizes an advanced technique for the absolute quantification of nucleic acids. Heralded as the third generation of PCR technologies, it is considered seminal in the arena of life sciences and clinical diagnostics. This concept was first brought to light in 1999 by luminaries Bert Vogelstein and Kenneth W. Kinzler.
Digital PCR is a unique system that micropartitions the samples into droplets, ensuring an average of one or zero target DNA molecules in each partition. Post PCR amplification, the presence or absence of fluorescence in discrete partitions is recorded using a binary system. Under this system, the presence of fluorescence is denoted as 1 and non-fluorescence as 0. Subsequent statistical analysis of the fluorescence signals yields an accurate absolute quantification of wild-type and mutant species within the testing sample. This high level of sensitivity allows for the detection of mutations as low as a single copy. (Figure 3)
Service you may intersted in
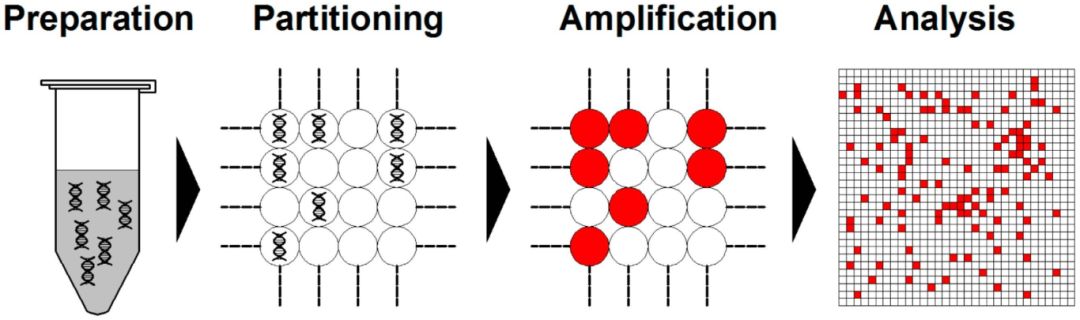
Figure.3 dPCR workflow (Cao et al., 2020)
3. HRM
High-Resolution Melting Curve (HRM) analysis, invented by Professor Carl Wittwer of Molecular Pathology at the University of Utah, is a technological advancement predicated on polymerase chain reaction (PCR); it's specially designed for gene mutation detection and genotyping. HRM primarily hinges on a system that faithfully combines PCR target fragments aptly designed for the purpose of use, a saturating DNA double-strand fluorescent dye, an instrument system capable of thermally-regulated high-resolution melting curve signal attainment, and a melting curve software analysis system. The technique, owing to its simplicity, cost-effectiveness, speed, closed-tube post-PCR mode that mitigates the risk of secondary contamination, and high detection sensitivity, has rapidly become widely adopted for various gene analyses in medical and other bio-related fields.
The main principle of HRM exploits the property of a saturating type of double-stranded DNA (dsDNA) dye capable of integrating into the PCR product. It involves real-time monitoring of dsDNA unwinding during the heating process, the subsequent detachments of the fluorescent dye leading to a reduction or total disappearance of fluorescent signals, and accurately recording the melting curve at high resolution. Thereby, allowing for a highly sensitive analysis of sample mutations, single nucleotide polymorphisms (SNPs), and methylation sites at the resolution of a single base.








